What exactly does No Hit mean in my analysis?
The RTL Genomics analysis pipeline generates a number of files as discussed in the section “Analysis Archive File Descriptions” on page 25. The taxonomic information given in the “FullTaxa” and “TrimmedTaxa” are often the assigned taxonomic information, but in some cases the taxonomic information may contain the “No Hit” keyword for each taxonomic level. The “No Hit” keyword simply means that there exists no matches in our database that meet the minimum criteria to be considered likely. As such we cannot make any confident calls regarding the taxonomic classification of the OTU – even at the Kingdom/Domain level. There exists a number of reasons why this occur which we will describe below:
The organism’s sequence is missing from our database
New sequences and organisms are added to NCBI/EMBL/DDBJ on a daily basis and, while RTL Genomics makes every effort to keep our databases current, it may take a few months before new sequences are in our database.
Many sequences in NCBI/EMBL/DDBJ are too short or contain no taxonomic information and are excluded from being added to our database.
The organism’s sequence data is not yet in NCBI/EMBL/DDBJ.
NCBI/EMBL/DDBJ contain a vast amount of data, however, they require researchers to have already sequenced an organism before they have sequence data to provide. If no one has sequenced the organism and submitted it to those repositories, then we will not yet have the sequence.
While this is not often the case, there exists the possibility that your sequence data contains an organism not yet known to science, i.e. a novel species
Low-quality sequence
While RTL Genomics does perform quality and chimera checking on your data, these algorithms are not fool-proof and low-quality or chimeric sequences may have managed to make it to the taxonomic analysis stage. These sequences will often fail to identify as any organism due to their low quality or chimeric nature, causing them to be marked as “No Hit”.
What is the difference between Unknown and Unclassified in my analysis files?
The RTL Genomics analysis pipeline generates a number of files as discussed in the section “Analysis Archive File Descriptions” on page 25. The taxonomic information given in the “FullTaxa” and “TrimmedTaxa” are often the assigned taxonomic information, but in some cases, the taxonomic information may contain the “Unknown” and “Unclassified” keywords which can cause some confusion. These keywords are described as follows:
Unknown
Our algorithm was unable to make a confident determination regarding the taxonomic classification at a certain level.
See section “USEARCH Global Search (Default)” on page 19 for more information on how we determine confidence.
Unclassified
The taxonomic information retrieved from NCBI contains missing information at this level.
For instance, if the best match in our database is classified in NCBI down to the Family level then our database will mark the Genus and Species as “Unclassified”.
How are confidence values determined?
The RTL Genomics analysis pipeline generates a number of files as discussed in the section “Analysis Archive File Descriptions” on page 25. Once each OTU has been aligned to our database, our algorithm will select the top/best six matches for the OTU and attempt to assign a confidence value to each taxonomic level. The top match is then compared against the other five to determine the number of matches at agree with the base match at each taxonomic level. The number of agreements is then converted into a confidence value using the following equation:
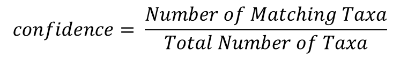
For instance, if OTU ID #13 has 6 top matches with the following taxa:
Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Bradyrhizobium sp
Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Bradyrhizobium sp
Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Bradyrhizobium sp
Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Bradyrhizobium japonicum
Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Bradyrhizobium japonicum
Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Rhodopseudomonas ; Rhodopseudomonas palustris
Then each taxonomic level would receive the following confidence:

Using this example, we would assign a confidence of 1 (100%) to the kingdom, phylum, class, order and family taxa. We would then assign a confidence of .83 (83%) to the genus taxon and a confidence of .5 (50%) to the species. These confidence values are then used when the “TrimmedTaxa” files are generated.
What is the difference between FullTaxa and TrimmedTaxa Files?
The RTL Genomics analysis pipeline generates a number of files as discussed in the section “Analysis Archive File Descriptions” on page 25. In our analysis data we provide most of our analysis in duplicate files, one containing the analysis using the “FullTaxa” and the other containing the analysis using the “TrimmedTaxa”. Please see “How are confidence values determined?” found on page 8 as this discussion will assume you have an idea of what confidence values are and how we assign them.
The FullTaxa files are generated under the assumption that the best match is the correct one. As such each taxonomic level is assigned using that match without taking the confidence values into account. We provide this file for two reason: 1) this method for assigning taxa is similar to the method we used many years ago and we continue to provide these files for legacy purposes and 2) this helps you see what the original best match was before confidence trimming occurred. We believe these files are a powerful tool in allowing you to better get an idea of what our algorithm originally thought the data contained before confidence values were taken into account, which can help you get a better feel for the data. However, we do advise that you do not use only the FullTaxa data to perform your analysis as the lack of confidence makes the data considerably less accurate.
The TrimmedTaxa files are generated using the FullTaxa data after the confidence values have been taken into account. As such each taxonomic level is assigned only if the confidence value is greater than or equal to .51 (51%). If a taxon falls below .51, it is replaced with the “Unknown” keyword. Using the example data provided in the section “How are confidence values determined?” on page 8, the FullTaxa would read Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Bradyrhizobium sp and the TrimmedTaxa would read Bacteria ; Proteobacteria ; Alphaproteobacteria ; Rhizobiales ; Bradyrhizobiaceae ; Bradyrhizobium ; Unknown, where the species is now assigned “Unknown” due to the low confidence value.
Brief Synopsis
Full Taxa
Generated under the assumption that the top/best match is completely accurate.
Does not take confidence values into account.
Provided primarily for legacy purposes and not recommended to be used for detailed analysis.
Trimmed Taxa
Takes the confidence value into account at each taxonomic level.
Replaces low confidence taxa with the “Unknown” keyword.
Do my sequences contain the primer, barcodes or adapters?
Upon the completion of an order at RTL Genomics, clients will receive two zip archives containing their sequence data, the files are described in the section “File Descriptions and Formatting” starting on page 20. The raw data archive contains your sequence data directly from the sequencer with no post-processing done on our end. This data is packaged as one SFF or one pair of FASTQ files per sample. The FASTA data archive contains your sequence data after we have performed denoising and some basic quality checking on the data. This data is provided as a single FASTA formatted sequence and quality file that contains all of your sequences multiplexed together.
Please refer to the following graph in order to determine whether the primer, barcodes, or adapters are on your sequences:
